
The NCSE, Judge Jones, and Citation Bluffs About the Origin of New Functional Genetic Information
[Editor’s Note: This article is adapted from a series of posts originally posted on Evolution News and Views. The originals may be seen here: Part 1, Part 2, Part 3, Part 4, Part 5, Part 6, Part 7, Part 8.]
I. Introduction
Not long before the beginning of the 2005 Kitzmiller v. Dover trial, then-National Center for Science Education staff member Nicholas Matzke told a reporter, “The origin of genetic information is thoroughly understood.”1 During the Dover trial, plaintiffs’ expert witness, biologist Kenneth Miller, testified that he presented Judge John E. Jones with “more than three dozen scientific studies showing the origin of new genetic information by these evolutionary processes.”2 The plaintiffs’ attorneys, working with the NCSE, successfully convinced Judge Jones to parrot Miller by stating in the Kitzmiller v. Dover ruling that Miller had “pointed to more than three dozen peer-reviewed scientific publications showing the origin of new genetic information by evolutionary processes.”3
Virtually all of those “publications” mentioned by Judge Jones came from one single paper Miller discussed at trial, a review article, co-authored by Manyuan Long of the University of Chicago.4 The article does not even contain the word “information,” much less the phrase “new genetic information.”5
The NCSE continues to cite Long et al. claiming that the origin of new functional biological information is nothing to be troubled about. In 2008, NCSE posted an online response to parts of Explore Evolution: The Arguments For and Against Neo-Darwinism, stating that “Biologists have no trouble showing how new information (in the sense used by information theorists) originates, nor how new genes, kinds of cells or tissues evolve.” Similar arguments appear in an article co-authored by former NCSE staff member Matzke critiquing critical analysis of evolution. Matzke writes with Paul Gross that it is “scandalously wrong” to argue that modern evolutionary biology has had difficulty accounting for the origin of new biological information because “[c]ompetent scientists know how new genetic information arises.”6 He too relies upon the paper by Long et al. asserting that “it reviews all the mutational processes involved in the origin of new genes and then lists dozens of examples in which research groups have reconstructed the genes’ origins.”7
But are Judge Jones’s, Ken Miller’s, and the NCSE’s bold proclamations supported? Does Long et al. actually reveal the origin of new biological information? Is Explore Evolution wrong? A closer look shows that the NCSE is equivocating over the meanings of the words “information” and “new,” and that the NCSE’s citations are largely bluffs, revealing little about how new genetic functional information could originate via unguided evolutionary mechanisms. This bluff was accepted at face value by Judge Jones, who incorporated it in his highly misguided legal ruling.
In fact the origin of new functional biological information is perhaps the most important question in biology. As origin of life theorist Bernd-Olaf Kuppers stated in his book Information and the Origin of Life, “The problem of the origin of life is clearly basically equivalent to the problem of the origin of biological information.”8
Judge Jones was not merely in error. Worse than any simple mistake, the misinformation he propounded in his ruling entered media and academic culture, becoming enshrined as a Darwinian myth, alongside many others. This myth holds that perhaps the most important question in biology has been solved, when really (as this article will show), that is far from being the case.
II. The Evolution-Lobby’s Useless Definition of Biological Information
For the NCSE / Ken Miller / Judge Jones to claim that there is an explanation or “the origin of new genetic information by evolutionary processes,” they must equivocate on the definitions of both the words “information” and “new.” Following the NCSE, Judge Jones probably would define information as “Shannon information,” which means mere complexity. Under this definition, a functionless stretch of randomly garbled junk DNA might have the same amount of “information” as a fully functional gene of the same sequence-length. For example, under Shannon information, which the NCSE would claim is “the sense used by information theorists,” the following two strings contain identical amounts of information:
String A:
SHANNONINFORMATIONISAPOORMEASUREOFBIOLOGICALCOMPLEXITYString B:
JLNUKFPDARKSWUVEYTYKARRBVCLTLODOUUMUEVCRLQTSFFWKJDXSOB
Both String A and String B are composed of exactly 54 characters, and each string has exactly the same amount of Shannon information—about 254 bits.9 Yet clearly String A conveys much more functional information than String B, which was generated using a random character generator.10 For obvious reasons, Shannon complexity has a long history of being criticized as an unhelpful metric of functional biological information. After all, biological information is finely-tuned to perform a specific biological function, whereas random strings are not. A useful measure of biological information must account for the function of the information, and Shannon information does not take function into account.
Some leading theorists recognize this point. In 2003, Nobel Prize winning origin of life researcher Jack Szostak wrote in a review article in Nature lamenting that the problem with “classical information theory” is that it “does not consider the meaning of a message” and instead defines information “as simply that required to specify, store or transmit the string.”11 According to Szostak, “a new measure of information – functional information – is required” in order to take account of the ability of a given protein sequence to perform a given function. Likewise, a paper in the journal Theoretical Biology and Medical Modelling observes:
[N]either RSC [Random Sequence Complexity] nor OSC [Ordered Sequence Complexity], or any combination of the two, is sufficient to describe the functional complexity observed in living organisms, for neither includes the additional dimension of functionality, which is essential for life. FSC [Functional Sequence Complexity] includes the dimension of functionality. Szostak argued that neither Shannon’s original measure of uncertainty nor the measure of algorithmic complexity are sufficient. Shannon’s classical information theory does not consider the meaning, or function, of a message. Algorithmic complexity fails to account for the observation that “different molecular structures may be functionally equivalent.” For this reason, Szostak suggested that a new measure of information—functional information—is required.12
In 2007 Szostak co-published a paper Proceedings of the National Academy of Sciences with Carnegie Institution origin of life theorist Robert Hazen and other scientists furthering these arguments. Attacking those who insist on measuring biological complexity using the outmoded tools of Shannon information, the authors wrote, “A complexity metric is of little utility unless its conceptual framework and predictive power result in a deeper understanding of the behavior of complex systems.” Thus they “propose to measure the complexity of a system in terms of functional information, the information required to encode a specific function.”13
Stephen C. Meyer follows this approach, writing in a peer-reviewed scientific paper that it is useful to adopt “‘complex specified information’ (CSI) as a synonym for ‘specified complexity’ to help distinguish functional biological information from mere Shannon information—that is, specified complexity from mere complexity.”14 Meyer’s suggested definition of “specified complexity” is useful in describing functional biological information. Specified complexity is a concept derived from the mainstream scientific literature and is not an invention of critics of neo-Darwinism. In 1973, origin of life theorist Leslie Orgel distinguished specified complexity as the hallmark of biological complexity:
[L]iving organisms are distinguished by their specified complexity. Crystals are usually taken as the prototypes of simple, well-specified structures, because they consist of a very large number of identical molecules packed together in a uniform way. Lumps of granite or random mixtures of polymers are examples of structures which are complex but not specified. The crystals fail to qualify as living because they lack complexity; the mixtures of polymers fail to qualify because they lack specificity.15
Orgel thus captures the fact that specified complexity, or CSI, requires both an unlikely sequence and a specific functional arrangement. Specified complexity is a much better measure of biological complexity than Shannon information, a point which the NCSE must resist because it’s much harder to generate specified complexity via Darwinian processes than mere Shannon complexity.
By wrongly implying that Shannon information is the only “sense used by information theorists,” the NCSE avoids answering more difficult questions like how the information in biological systems becomes functional, or in its own words, “useful.” Rather, the NCSE seems more interested in addressing simplistic, trivial questions like how one might add additional characters to a string, or duplicate a string, without regard for the all important question of whether those additional characters convey some new functional message. Since biology is based upon functional information, Darwin-skeptics are interested the far more important question of, Does neo-Darwinism explain how new functional biological information arises?
III. The Evolution-Lobby’s Misguided Definition of “New”
When Judge Jones claimed that Ken Miller showed “the origin of new genetic information by evolutionary processes,” Ken Miller and his friends at the NCSE didn’t just equivocate on the definition of “information,” they also misused the term “new.” In fact, they would likely accept something as “new” if it were merely a copy or a duplicate some pre-existing stretch of DNA, even if the new copy doesn’t actually do anything new, or perhaps even when the new DNA doesn’t do anything at all. In contrast, proponents of intelligent design would define “new” genetic information as a new stretch of DNA which actually performs some different, useful, and new function. For example, consider the following string:
DUPLICATINGTHISSTRINGDOESNOTGENERATENEWCSI
This 42-character string has ~197 bits of Shannon information. Now consider the following string longer:
DUPLICATINGTHISSTRINGDOESNOTGENERATENEWCSIDUPLICATINGTHISSTRINGDOESNOTGENERATENEWCSI
This procedure just added 42 “new” characters, but no new function has been produced. Assuming there was no way to predict beforehand that the first string would be duplicated just as it was, the amount of Shannon information has doubled, but the amount of CSI has not increased one bit (literally).
The above example is of course analogous to the commonly cited evolutionary mechanism of gene duplication, which evolutionists commonly cite as a mechanism by which Darwinian processes can produce new information. But new functional information is not generated by a process of duplication until mutations change the gene enough to generate a new function—which may or may not be possible. As Professor of Neurosurgery Michael Egnor insightfully said in response to one evolutionary biologist:
[G]ene duplication is, presumably, not to be taken too seriously. If you count copies as new information, you must have a hard time with plagiarism in your classes. All that the miscreant students would have to say is ‘It’s just like gene duplication. Plagiarism is new information- you said so on your blog!’16
Indeed, evolutionary explanations cannot simply rely upon duplication, for there must be duplication followed by recruitment to a new function. However one defines “information,” merely duplicating a string does not produce new functional information.17
IV. Finding Darwin in All the Wrong Places
Despite the fact that proponents of neo-Darwinian evolution claim to understand the origin of new genetic information, they obscure the fact that they lack explanations for such by making vague appeals to mechanisms such as “gene duplication,” “rearrangement,” and “natural selection.” Such mechanisms are generally inferred from circumstantial evidence, i.e. similarities and differences between gene sequences, where a neo-Darwinian evolutionary history is assumed. More importantly, accounts that invoke such mechanisms almost never attempt to assess the likelihood of mutations producing the genetic changes in question. In this regard, important notes of caution must be observed when assessing evolutionary accounts of the origin of a gene.
A 2007 article by evolutionary biologist Michael Lynch in Proceedings of the National Academy of Sciences USA goes to the heart of some of the assumptions inherent in many claims of neo-Darwinian evolution. Lynch provides a list of myths promoted by biologists, and he calls it a “myth” to believe that “Characterization of interspecific differences at the molecular and/or cellular levels is tantamount to identifying the mechanisms of evolution.”18
Of course, one of the typical “mechanisms of evolution” cited is natural selection, commonly invoked to account for how a gene duplicate acquires a new function. But what kind of evidence is sufficient to demonstrate that positive selection, or natural selection acting to preserve adaptive mutations, has occurred? Biologist Austin Hughes warns that most inferences of positive selection are based upon questionable statistical analyses of genes:
A major hindrance to progress has been confusion regarding the role of positive (Darwinian) selection, i.e., natural selection favoring adaptive mutations. In particular, problems have arisen from the widespread use of certain poorly conceived statistical methods to test for positive selection. Thousands of papers are published every year claiming evidence of adaptive evolution on the basis of computational analyses alone, with no evidence whatsoever regarding the phenotypic effects of allegedly adaptive mutations. … Contrary to a widespread impression, natural selection does not leave any unambiguous ‘‘signature’’ on the genome, certainly not one that is still detectable after tens or hundreds of millions of years. To biologists schooled in Neo-Darwinian thought processes, it is virtually axiomatic that any adaptive change must have been fixed as a result of natural selection. But it is important to remember that reality can be more complicated than simplistic textbook scenarios. … In recent years the literature of evolutionary biology has been glutted with extravagant claims of positive selection on the basis of computational analyses alone … This vast outpouring of pseudo-Darwinian hype has been genuinely harmful to the credibility of evolutionary biology as a science.19
In short, evolutionary biologists commonly assume that mutations that change protein sequence were fixed by natural selection, but this assumption may not hold true since many such mutations are neutral and confer no selective advantage.
Biochemist Michael Behe offers another reason not to infer neo-Darwinian mechanisms of change based upon mere evidence of sequence similarity:
Although useful for determining lines of descent … comparing sequences cannot show how a complex biochemical system achieved its function—the question that most concerns us in this book. By way of analogy, the instruction manuals for two different models of computer put out by the same company might have many identical words, sentences, and even paragraphs, suggesting a common ancestry (perhaps the same author wrote both manuals), but comparing the sequences of letters in the instruction manuals will never tell us if a computer can be produced step-by-step starting from a typewriter. … Like the sequence analysts, I believe the evidence strongly supports common descent. But the root question remains unanswered: What has caused complex systems to form?20
[M]odern Darwinists point to evidence of common descent and erroneously assume it to be evidence of the power of random mutation.21
Many scientific papers purporting to show the evolution of “new genetic information” do little more than identify molecular similarities and differences between existing genes and then tell evolutionary just-so stories of duplication, rearrangement, and subsequent divergence based upon vague appeals to “positive selection” that purport to explain how the gene arose. But exactly how the gene arose is never explained. In particular, whether chance mutations and unguided natural selection are sufficient to produce the relevant genetic changes is almost never assessed.22 These scientific papers—especially the citation bluffs offered by the NCSE / Judge Jones / Ken Miller—play the Gene Evolution Game, an easy game to play which ultimately tells us little about the origin of new functional genetic information, as we’ll see the next section.
V. How to Play the Gene Evolution Game (note: this section is written tongue-in-cheek)
The Gene Evolution Game is a very simple game to play. In three examples, we’ll develop three rules that can help you explain the origin of any new gene. That’s right—any gene! Let’s start with a simple example:
Rule 1: The Magic Wand of Gene Duplication
Where do new genes come from? Gene duplication is typically how we explain where a new gene comes from. Here’s how it works:
- Take a gene you’ve observed in some organism. We’ll call it Gene B.
- Find another gene similar to Gene B. Let’s call it Gene A.
- Claim that at some time in the past, Gene A duplicated so then there were two copies of Gene A.
- Then assert that one of Gene A’s duplicates evolved into Gene B.
Gene duplication is thus very a powerful explanation, and it looks like this:
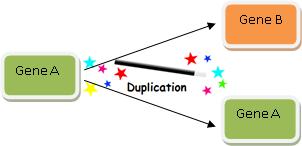
Wasn’t that easy? We’ve just explained how Gene B evolved! So when you find two genes with high sequence similarity, you can always explain how one evolved from the other via the magic wand of Gene Duplication.

The NCSE says “Gene duplication are [sic] common events, resulting from small errors in the process of cell replication. Once a gene is duplicated it is possible for one copy to mutate, adding information without risking the functioning of the pre-existing gene.” That’s all you need to know—when you invoke duplication, you needn’t worry about whether there is some functional evolutionary pathway for the duplicate gene to follow as it acquires some new function. In other words, you don’t need to worry about how new functional genetic information arises because “gene duplication” explains everything worth explaining! It’s easy to get extra genetic information in the Shannon sense, and that’s all that matters.
Rule 2: No Worries—Natural Selection Can Do It!
Now obviously the modern version of Gene B we find doesn’t perfectly resemble Gene A, or else it would be Gene A. So we have to account for how a copy of Gene A acquired its new function—Function B. One might think this would be the key part of explaining how new functional genetic information arises, but believe it or not, this is actually the easiest and quickest aspect of the game: we just call upon the power of “natural selection” and the problem is solved! This diagram shows exactly how we do it:
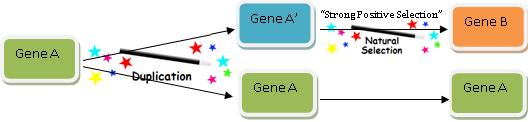
The great thing about the Gene Evolution Game is that natural selection can change (or not change, depending on what you wish) almost anything. And I mean anything.
Don’t worry about the details. If you want to account for differences between Gene B and Gene A, natural selection is always up to the challenge. Don’t worry about whether Gene A’ could evolve from Function A to Function B by small sequential adaptive steps. Don’t worry about the order in which amino acids changed, or whether many mutations were necessary to gain any functional advantage (that sort of thing is too unlikely to occur anyway, so just ignore it). Don’t worry about adaptive constraints, weak selection, or loss due to genetic drift. And most of all, definitely don’t do any calculations to determine the likelihood of whether all of the changes could have occurred in any reasonable amount of time.
We know the gene must have evolved, therefore it did evolve. Thus, you can think of natural selection as another magic wand. It may be invoked at any time to explain how a gene changed or evolved to acquire its new function.

This wand is a very powerful tool—it can explain both why things change, and why things stay the same.23 Wow!
Rule 3: The Magic Wand of “Rearrangement”
To play the Gene Evolution Game, there’s one last trick you need to know. Sometimes Gene B isn’t similar to just Gene A. Sometimes part of Gene B looks like Gene A, but another part looks like another gene. We’ll call the latter Gene Z. Don’t worry—this is all still easy to explain! We start by invoking duplication: Imagine that Gene A and Gene Z both duplicated, and then both duplicate copies were suddenly transported across the genome so that now they reside on a chromosome right next to one another. This is called “rearrangement.” If this sounds a little complicated, we’ll draw some diagrams to show how it works:
Step 1: Gene A and Gene Z are each in different locations, maybe even on different chromosomes:
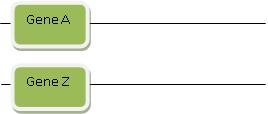
Then a special process called “rearrangement” suddenly rearranges and transports Gene A and Gene Z so they’re right next to each other in some other location in the genome. Rearrangement is a powerful magic wand you can invoke to explain how two stretches of DNA that initially are far apart suddenly end up near one another. They then can form a new functional gene. You’re probably getting a feel by now for how this works:

There are all kinds of rearrangements you can invoke—insertions, deletions, inversions, translocations—and you can invoke them in virtually any order and in any amount you please to explain how you get any two, or three, or even dozens of pieces of DNA to come together from throughout the genome to end up right next to one-another so that presto, you have your new functional gene. Just mix and match these types of rearrangements as needed to create virtually whatever DNA sequence you desire—rearrangement is always up to the task.

It’s all downhill from here. Natural selection can then perfect the rearranged gene to make it functional. Never mind detailed demonstrations that this actually works. Just sprinkle some natural selection and Gene A and Gene Z will magically combine functions and evolve into Gene B. Here we go, completing the explanation with everything we need to know:

Using the three magic wands of duplication, rearrangement, and natural selection you can provide a full and complete detailed explanation for the evolution of virtually any gene.
No Identifiable Ancestor? No Worries!
First, in some cases, your gene (i.e., Gene B) only has a homologue known from an entirely different species. So how did Gene B arrive in your organism? In these cases, just invoke lateral gene transfer (LGT) to whoosh the right gene into your organism. It doesn’t even matter whether lateral gene transfer is thought to occur between the organisms you’re working with—if the gene you need is found in some other species, then that by itself is evidence that lateral gene transfer occurs between the organisms you’re working with!24
Second, sometimes part of your gene doesn’t resemble part of any other known gene anywhere. Some people might wonder, “Where did this gene come from?” You still don’t worry about this. Remember what we said about natural selection? It can change anything. So if you can’t find any similar genes, just assume that your unique DNA sequence has evolved so much due to natural selection that it just doesn’t resemble its ancestral sequence any longer. But don’t worry, it’s not, and never is, the case that there wasn’t an ancestor. It’s just that the strong powers of natural selection changed the gene so much that we can’t identify any possible ancestral sequence.25
Some Final “Do’s” and “Don’ts” of the Gene Evolution Game
Right about now, you might be wondering about that last example we gave. So before you go any further, here’s a reminder of some questions you don’t need to ask:
- Given the known effects and rates of mutations, what were the odds of Gene A and Gene Z suddenly being rearranged next to one-another so that they could now function together as one single new gene product, Gene B?
- Did the rearranged gene product B start out functional? If not, how quickly could it gain function? How was it preserved from loss until it became functional?
- Are proteins really as malleable as this story would suppose or would the new combined gene encounter folding or other contextual problems?
- What mutational pathway was taken to evolve Gene A and Gene Z into a new gene with function B?
- What selective advantages were gained at each small step of this evolutionary pathway?
- Were any “large steps” (i.e., multiple specific mutations) ever required to gain a selective advantage along the evolutionary pathway? Would such “large steps” be likely to occur?
- Could all of this happen on a reasonable timescale?
You don’t need to worry about these questions. In fact, believe it or not, you don’t even need to know the function of your gene to claim it evolved from A and Z! All you need to know is that Genes A, Z, and B exist. This summary of these 3 simple rules of the Gene Evolution Game will help you explain anything:
Gene Evolution Game Rule 1: Whenever you find sequence homology between two genes, just invoke a duplication event of some hypothetical, ancient ancestral gene, and you can explain how two different genes came to share their similarities.
Gene Evolution Game Rule 2: When you need to explain how a gene acquired some new function, or evolved differences from another gene, just invoke the magic wand of natural selection. No need to demonstrate that there is any benefit to the new gene, or that a step-wise path to adaptation exists. Finally, natural selection is especially useful when part of your gene appears unique—since natural selection can change anything, just conclude that natural selection changed your gene so much that it no longer resembles its ancestor.
Gene Evolution Game Rule 3: When a gene seems to be composed of the parts of several genes, just invoke duplications and rearrangements of all the DNA sequences you need, so you can get them all together in the right place. If you need to delete parts of a gene, or invert them, or transpose to a new location, just invoke different types of rearrangements as often and as liberally as you wish, and ba-da-bing, you’ve got your new gene!
And remember, don’t ask those other hard questions. Just use these three rules and you can explain virtually anything. No details required!
VI. Asking the Right Questions about the Evolutionary Origin of New Biological Information
In all seriousness, we’ve seen above that it’s easy to duplicate a gene, but the key missing ingredient in many neo-Darwinian explanations of the origin of new genetic information is how a gene duplicate then acquires some new optimized function. Evolutionists have not demonstrated, except in rare cases, that step-wise paths to new function for duplicate genes exist.
As discussed above, Austin Hughes cautions against making “statistically based claim[s] of evidence for positive selection divorced from any biological mechanism.”26 In other words, natural selection is invoked to explain the evolution of genes where we do not even know the functional effect of the mutations being asserted. In this regard, Hughes observes that even in one of the more sophisticated studies, “there was no direct evidence that natural selection was actually involved in fixing adaptive changes.”27
Hughes also acknowledges a problem inherent in many appeals to natural selection, namely that required mutations may not give any selective advantage when they first arise. He thus writes regarding one study:
For example, a rhodopsin from the Japanese conger eel with λmax ≈ 480 nm achieved this sensitivity through the interaction of three different amino acid replacements (at sites 195, 195, and 292). There does not seem to be any way that natural selection could favor an amino acid replacement that would be of adaptive value only if two other replacements were to occur as well.28
In this case, there was no stepwise advantage gained with each successive mutation. Because no advantage could have been gained until all three mutations were present, Hughes finds it more “plausible” to believe that the first two mutations were “selectively neutral” and became fixed due to random, non-adaptive processes such as genetic drift. Once the third mutation arose it might have provided an advantage, but to paraphrase Scott Gilbert, at best this really only explains the survival of the fittest, not the arrival of the fittest.29
But Hughes’ explanation has deep deficiencies: it requires that two mutations become fixed before any selective advantage for the third mutation is gained. This implies that there must be three specific mutations to gain any selective advantage. A key question is thus, Are multiple specific mutational changes likely to appear in the same individual through unguided chance mutations given known mutation rates and population sizes? Even Hughes, despite his exhortations to fellow evolutionary biologists to employ more rigor in their studies, does not address this fundamental question.
A similar example is found when leading paleoanthropologist Bernard Wood critiqued a simplistic model of human cranial evolution on the grounds that too many mutations would be required to gain any functional advantage:
The mutation would have reduced the Darwinian fitness of those individuals. . . . It only would’ve become fixed if it coincided with mutations that reduced tooth size, jaw size and increased brain size. What are the chances of that?30
Similarly, Jerry Coyne writes that “It is indeed true that natural selection cannot build any feature in which intermediate steps do not confer a net benefit on the organism.”31 This highlights a key deficiency in many neo-Darwinian accounts of the evolution of genes. Namely, they fail to demonstrate that the processes necessary to generate new functionally advantageous genetic information are plausible. As with Hughes’s or Wood’s examples above, multiple mutations might be necessary to gain any functional advantage. Any account invoking blind, unguided, random mutations to evolve a gene from Function A to Function B must address at least these three questions:
- Question 1: Is there a step-wise adaptive pathway to mutate from A to B, with a selective advantage gained at each small step of the pathway?
- Question 2: If not, are multiple specific mutations ever necessary to gain or improve function?
- Question 3: If so, are such multi-mutation events likely to occur given the available probabilistic resources?
Mathematician David Berlinski considers such questions when critiquing evolutionary accounts of eye evolution. Darwinian processes fail because multiple changes are required for a new function to appear:
If these changes come about simultaneously, it makes no sense to talk of a gradual ascent of Mount Improbable. If they do not come about simultaneously, it is not clear why they should come about at all.32
Again, the key question is therefore, how hard is it for new functional biological information to arise? Answering this question requires assessing the ability of random mutation and natural selection to generate new functional biological information. But when most evolutionary biologists play the Gene Evolution Game, they do not make such assessments and rarely consider these questions. Instead they typically invoke processes such as gene duplication, natural selection, and rearrangement, without demonstrating that random and unguided mutations are sufficient to produce the information needed. Any explanation that at base is little more complicated than “duplication, rearrangement, and natural selection” is not a demonstration that new functional genes can arise by unguided processes.
Thankfully, some scientists are willing to consider these key questions. They have performed research providing data that offers strong reasons to be skeptical of the ability of mutation and selection to form new functional genetic sequences.
A. Asking Questions 1 and 2:
Molecular biologist Doug Axe has performed mutational sensitivity tests on enzymes and found that functional protein folds may be as rare as 1 in 1077.33 His research shows that the fitness landscape for many enzymes looks like this, making it very unlikely that neo-Darwinian processes will find the specific amino acid sequences that yield functional protein folds:

To put the matter in perspective, these results indicate that the odds of Darwinian processes generating a functional protein fold are less than the odds of someone closing his eyes and firing an arrow into the Milky Way galaxy, and hitting one pre-selected atom.34 To say the least, this exhausts the probabilistic resources available. Such data help us answer the first question: it’s not likely that there will be a functional stepwise mutational pathway leading from Function A to Function B.
Douglas Axe is by no means the only biologist to make this observation. A leading college-level biology textbook, Campbell’s Biology, observes that “Even a slight change in primary structure can affect a protein’s conformation and ability to function.”35 Likewise, David S. Goodsell, an evolutionist biologist, writes:
As you might imagine, only a small fraction of the possible combinations of amino acids will fold spontaneously into a stable structure. If you make a protein with a random sequence of amino acids, chances are that it will only form a gooey tangle when placed in water. Cells have perfected the sequences of amino acids over many years of evolutionary selection…36
What Goodsell does not mention is that if “perfected” amino acid sequences and functional protein folds are rare and slight changes can disrupt function, then selection will be highly unlikely to take proteins from one functional fold to the next without traversing some non-functional stage. So how do new functional protein folds evolve? This effectively answers question two, implying that many specific mutations would be necessary for evolving genes to pass through non-functional stages while evolving some new function. Question 3 assesses whether this is likely to happen.
B. Asking Question 3:
In 2004, Michael Behe and physicist David Snoke published a paper in the journal Protein Science reporting results of computer simulations and theoretical calculations. They showed that the Darwinian evolution of a simple functional bond between two proteins would be highly unlikely to occur in populations of multicellular organisms. The reason, simply put, is because too many amino acids would have to be fixed by non-adaptive mutations before gaining any functional binding interaction. They found:
The fact that very large population sizes—109 or greater—are required to build even a minimal [multi-residue] feature requiring two nucleotide alterations within 108 generations by the processes described in our model, and that enormous population sizes are required for more complex features or shorter times, seems to indicate that the mechanism of gene duplication and point mutation alone would be ineffective, at least for multicellular diploid species, because few multicellular species reach the required population sizes.37
According to this data, chance mutations are unlikely to produce even two required non-adaptive mutations in multicellular diploid species within any reasonable timescale. This answers the third question: getting multiple specific non-adaptive mutations in one individual is extremely difficult, and more than two required but non-adaptive mutations are likely beyond the reach of multi-cellular organisms. Studies like this show that the actual ability of random mutation and unguided selection to produce even modestly complex new genetic functions is insufficient.
In 2008, Behe and Snoke’s would-be critics tried to refute them in the journal Genetics, but found that to obtain only two specific mutations via Darwinian evolution “for humans with a much smaller effective population size, this type of change would take > 100 million years.” The critics admitted this was “very unlikely to occur on a reasonable timescale.”38 In other words, there is too much complex and specified information in many proteins and enzymes to be generated in humans by Darwinian processes on a reasonable evolutionary timescale.
As noted in the comments on the Gene Evolution Game, when neo-Darwinists try to explain the evolution of genes, mere point mutations often are insufficient to account for the gene’s sequence. They must therefore appeal to genetic rearrangements such as insertions, deletions, or an alleged process called “domain shuffling” where segments of proteins become shuffled to new positions in the genome. In his book The Edge of Evolution, Michael Behe reviews research that engineered new protein function by swapping domains to change protein function, and found that the intelligently engineered changes required multiple modifications that, in nature, would require too many simultaneous mutational events to yield functional changes:
[Protein engineering research] does not mimic random mutation. It is the exact opposite of random mutation. … What do the lab results tell us about whether random-yet-productive shuffling of domains “occurs with significant frequency under conditions that are likely to occur in nature”? About whether that is biologically reasonable? Nothing at all. When a scientist intentionally arranges fragments of genes in order to maximize the chances of their interacting productively, he has left Darwin far, far behind. … [Experiments that engineered proteins by shuffling domains] didn’t just splice two genes together in a single step; they took several additional steps as well. … Remember the more steps that have to occur between beneficial states, the much less plausible are Darwinian explanations. … Domain shuffling would be an instance of the “natural genetic engineering” championed by James Shapiro where evolution by big random changes is hoped to do what evolution by small random changes can’t. But random is random. No matter if a monkey is rearranging single letters or whole chapters, incoherence plagues every step. … One step might luckily be helpful on occasion, maybe rarely a second step might build on it. But Darwinian processes in particular and unintelligent ones in general don’t build coherent systems. So it is biologically most reasonable to conclude that, like multiple brand new protein-protein binding sites, the arrangement of multiple genetic elements into sophisticated logic circuits similar to those of computers is also well beyond the edge of Darwinian evolution.39
As Behe observes, “No matter if a monkey is rearranging single letters or whole chapters, incoherence plagues every step.” Thus, when multiple mutational events—whether point mutations, “domain shuffling,” or other types of rearrangements—are required to gain some functional advantage, it seems unlikely that blind neo-Darwinian processes can produce the new biological function.
Unfortunately, few if any advocates of the neo-Darwinian just-so stories investigate whether mutation and natural selection are sufficient to produce new functional genetic information. Instead they believe that finding similarities and differences between genes demonstrates that neo-Darwinian evolution has occurred, and they assume that “positive selection” is a sufficient explanation.
As Hughes cautions, they engage in “use of certain poorly conceived statistical methods to test for positive selection,” causing “the literature of evolutionary biology [to become] glutted with extravagant claims of positive selection” resulting in a “vast outpouring of pseudo-Darwinian hype [that] has been genuinely harmful to the credibility of evolutionary biology as a science.”40 Or, as Michael Behe cautions, they confuse mere sequence similarity with evidence of neo-Darwinian evolution. Finally, Michael Lynch warns his colleagues that “Evolutionary biology is not a story-telling exercise, and the goal of population genetics is not to be inspiring, but to be explanatory.”41
With these principles in mind, we’ll now assess about a dozen of the just-so stories concerning the origin of genes offered in studies cited by the NCSE.
VII. Assessing the NCSE’s Citation Bluffs on the Evolution of New Genetic Information
During the Kitzmiller v. Dover trial, Judge Jones followed Ken Miller and the NCSE by citing a review article in Nature Reviews Genetics co-authored by Manyuan Long.42 Jones claimed the paper shows “peer-reviewed scientific publications showing the origin of new genetic information by evolutionary processes.”43 In fact, what Long et al. actually demonstrates is that neo-Darwinists do not want to ask the right questions — the hard questions — about the sufficiency of their theory to explain gene evolution. They accept superficial just-so stories in place of detailed, plausibly demonstrated explanations.
Just as in the “Gene Evolution Game,” the studies cited in the review by Long et al. repeatedly invoke gene duplication, natural selection, and genetic rearrangements. But many offer little more than vague just-so stories that commit the mistakes Lynch warns of — mistaking story-telling for explanation.
To show how heavily the NCSE relies on Long et al. in its response to Explore Evolution, let’s look at how the NCSE reproduces a lengthy table (Table 2) from Long et al. The table lists a number of genes whose evolutionary origin has supposedly been explained.44 Many of the examples from this Table 2 are mere story-telling exercises based upon assumptions which do not explain or answer deeper questions about how neo-Darwinian evolution generates new functional genetic information:
a. Jingwei
The first entry in the table comes from a study that Long co-authored with Charles Langley in Science. The study asserts that a fruit fly gene, jingwei, arose when part of another gene, Adh, was retrotransposed into a new location on a fruit fly chromosome near a duplicate of the gene yellow-emperor.45 Their evidence for this rearrangement is sequence similarity between part of jingwei and Adh, and part of jingwei and yellow-emperor. Thus, invoking Gene Evolution Game Rules 1 and 3, the authors tell a story that presumes that hypothetical duplicates of yellow-emperor and Adh were fortuitously spliced together to create a new functional gene—jingwei. The exact word used is that exons were “recruited” from elsewhere into the genome “by capturing several upstream exons and introns of an unrelated gene” to produce “a new functional gene.” They author make no attempt to address the more important questions, such as whether a step-wise path to such a genomic rearrangement could have happened by unguided chance to fortuitously produce this gene. Merely finding sequence similarity between exons and other genes (or pseudogenes) does not thereby demonstrate neo-Darwinian evolution.
Long et al. claim that jingwei is only 2.5 million years old, but the original study compared the Adh-like exon in jingwei with the allegedly ancestral exon from Adh and found that they were so different that they must have diverged at least 30 million years ago. This poses a problem, because this fruit fly clade is not thought to be nearly that old; as Long and Langley write, “This conflicts with the age of the melanogaster subgroup, which is estimated to be 17 to 20 million years.” More important, the unexpectedly high degree of differences between the exons is taken, under neo-Darwinian assumptions, as evidence that jingwei “responded to positive natural selection and evolved a new function.” Yet according to one commentator, despite the fact that they are sure natural selection drove this gene to acquire its new function, “its actual function is obscure.”46 So they claim that natural selection was the driving mechanism, but they do not even know for sure in this paper that the gene has a function. They have not addressed any of the deeper questions of gene evolution, instead offering an incomplete and assumption-based story that ignores warnings from Austin Hughes against invoking “positive selection divorced from any biological mechanism.”47
b. Sdic
A second study cited by Table 2 asserts that various genes were duplicated, parts of which were then fused to create a new gene “de novo.”48 The authors wanted to explain how part of one gene, Cdic, became fused with part of another gene, Annx, but they ran into problems because the genes exist on the chromosome in a different order from the gene being studied. Making complicated use of Rules 1 and 3 of the Gene Evolution Game, they speculate that there was a series of duplications and rearrangements—highly selective and specific deletions—and then more duplications to produce this gene. This included one non-coding region spontaneously becoming a coding region, termed the de novo origin of a gene. After this complicated story, the paper concludes that Sdic arose from “extensive refashioning” of the genome.
First, although a testes-specific promoter was essential for Sdic, this unusual regulatory region did not really “evolve.” Instead it was aboriginal, created de novo by the fortuitous juxtaposition of suitable sequences. The more extensive evolutionary changes took place in Cdic intron 3, enabling an originally untranslatable sequence to become a new coding region whose product functions in the assembly of axonemal dynein.49
This “de novo” origin of a functional gene is an event that even Long et al. admits is “rare.”50 The authors then invoke strong positive selection due to the unlikelihood that such a dramatic reorganization “would have originated and been maintained in the absence of positive selection.” Despite their appeal to positive selection, the authors admit they aren’t even sure exactly what the gene does, stating: “We do not yet know how Sdic contributes to the function of the sperm axoneme, or even whether it is essential for male fertility.” So once again, they are sure it evolved due to “positive selection” but they do not even know exactly what function was being selected for.
A gene’s being “created de novo by the fortuitous juxtaposition of suitable sequences,” a mechanism that is “rare,” is not a compelling evolutionary explanation. This incomplete just-so story vaguely appeals to multiple mutations without assessing whether they would be likely to occur or what advantage they are offering. The story is no explanation at all.
c. Cid
The authors of this paper studied nucleotide differences between Cid genes in two closely related fruit fly species and found that nucleotide differences that led to changes in amino acid sequence were nearly 10 times more common than “silent” differences that did not affect amino acid sequence.51 Using Darwinian assumptions and Gene Evolution Game Rule 2, this led the authors to conclude that there was positive selection pressure on the gene to evolve.
Yet in this study natural selection was invoked not only to explain how genes changed, but also how genes stayed the same: a low number of replacement changes were taken as evidence of a “selective sweep,” a strong purifying selection that weeded out variation, to prevent change in one lineage. Thus, both a high degree of amino-acid changing differences and a low degree of amino-acid changing differences were taken as evidence of natural selection. Whether any of this is correct is purely a matter of ad hoc inference and starting assumptions. Moreover, the authors provided no mutation-by-mutation account to explain the selective advantages (or lack therefore) that might have been generated by any amino acid changes.
In light of the study’s methodology, Michael Lynch’s warning now comes to mind. It is a “myth” to believe that “[c]haracterization of interspecific differences at the molecular and/or cellular levels is tantamount to identifying the mechanisms of evolution.” Additionally, this study violates Austin Hughes’s admonition against “the widespread use of certain poorly conceived statistical methods to test for positive selection” which have caused “the literature of evolutionary biology [to become] glutted with extravagant claims of positive selection on the basis of computational analyses alone” resulting in a “vast outpouring of pseudo-Darwinian hype [that] has been genuinely harmful to the credibility of evolutionary biology as a science.”52 It’s also noteworthy that this study merely investigated how variations of the same gene originated in two closely related species, not how a new gene originated in the first place.
d. Arctic AFGP and Antarctic AFGP
Two papers cited by Table 2 in Long et al. discuss the origin of antifreeze genes (AFGP) in species of Arctic and Antarctic fish. The two species have similar antifreeze genes, even though they exist on literally opposite sides of the globe and are only distantly related. For the neo-Darwinist, these findings require that “near-identical antifreeze glycoproteins”53 evolved independently in distantly related species of fish—one in the Arctic and another in the Antarctic—via what is called “a striking case of convergent evolution.”54
Employing Gene Evolution Game Rules 1 and 3, a paper commenting on this research states the genes arose by “[d]uplication, divergence, and exon shuffling” and were “cobbled together from DNA of no related function (or no function at all).”55 For key parts of the antifreeze gene in Arctic cod, the commentators noted that the investigators “did not find any database matches to the sequence”56 and therefore could not determine its origin. However, there were matches for the Antarctic AFGP sequence, where similarities were found with part of a trypsinogen gene. This led to speculation about an evolutionary scheme that started with a trypsinogen gene, most of which was then deleted, followed by “recruitment” of a short threonine-alanine-alanine coding element, which then led to “de novo amplification of a short DNA sequence to spawn a novel protein with a new function.”57 This “de novo amplification of the coding element gave rise to an entirely new coding region that encodes the repetitive tripeptide backbone of AFGP,” even though this key component had “arisen (in part) from noncoding DNA.”58 Thus, according to their story, non-coding DNA spontaneously became functional and was duplicated many times to create the core functional “backbone” of this gene. No attempt was made to assess the mutational odds of such DNA that has “no function at all” suddenly becoming a key functional component of this gene.
This evolutionary story also solves problems through vague appeals to Gene Evolution Game Rule 2. The many genetic changes necessary to suddenly create this functional antifreeze gene were apparently accounted for by simply appealing to “powerful environmental selectional pressure” due to the need of the fish to survive in cold water.59 Of course, no statistical analyses were performed to assess the likelihood of cobbling together functional genes from completely unrelated stretches of DNA, some of which was previously non-functional, to produce a new functional antifreeze gene. Rather, one paper simply asserted the “creative” power of “molecular mechanisms”:
To consider the AFGP story as a special case of duplication and divergence would be oversimplifying; it is clear that the antifreeze function, or even a related function that could be converted to the purpose, was not present in trypsinogen. The molecular mechanisms involved in the formation of this gene were indeed more creative—making sense from nonsense—by calling into a functional coding capacity intronic DNA sequences.60
Are these molecular mechanisms likely to produce this gene? Are random mutations likely to “mak[e] sense from nonsense”? No analysis was given. The antifreeze genes are polyproteins, meaning they are complex many-in-one proteins designed to be cut into many pieces of specific lengths, each of which performs an important antifreeze function. The different segments are separated by special separator markers and cleaved by a specific protease. In this regard, no analysis was given to account for the origin of associated cleaver protease enzymes necessary for the function of the AFGP gene.
These papers base their claims of evolution purely upon circumstantial evidence—comparisons of sequence similarity—and then tell a tale of deletion, reshuffling, and amplification. Explanation of these genes by “cobbling” via “[d]uplication, divergence, and exon shuffling” and “de novo” recruitment of non-coding sequences does not account for how such a complex gene could actually originate. This story does not address how the complex many-proteins-in-one nature of these proteins evolved, nor was any consideration given the odds of spontaneously producing this functional gene. Nor have these investigators explained the highly unlikely event that two species would independently evolve highly similar antifreeze proteins.
The antifreeze proteins are highly repetitive, and may have less specified complexity than most proteins. Nonetheless, there’s no real evidence for neo-Darwinian evolution here, only sequence comparisons and a lot of missing details.
e. Adh-Finnegan
This article cited by Long et al. represents an example where a stretch of DNA that was previously presumed to be a “nonfunctional” pseudogene turned out to be a functional gene.61 The functional gene was then named Adh-Finnegan after “Tim Finnegan, a character from an Irish folksong, [who] was mistakenly declared dead and subsequently arose during his own wake.” This is a good example of how the junk-DNA myth initially led scientists to the wrong conclusion about this gene.
This paper’s just-so story makes use of all three rules of the Gene Evolution Game. Despite its citation in Long et al. (and thus by the NCSE), the study sheds very little light on the origin of the gene in question, other than to claim it evolved from another highly similar Adh gene and then “recruited” sequences via rearrangement from elsewhere in the genome. Predictably, an ancient duplication event is invoked to account for the origin of the gene, and then selection is invoked as a magic wand to account for “radical change in the structure” of the gene “compared to that of its highly conserved Adh ancestor.”
Extensive rearrangements are also invoked to explain how the gene “recruited ~60 new N-terminal amino acids,” as well as “the acquisition of new amino acid residues upstream from the ancestral ATG initiation codon.” The origin of the N-terminal exon posed a problem, however, because “A database search revealed no similarity of the N-terminal exon to known proteins,” and thus as Long et al. note, the gene must have “[r]ecruited a peptide from an unknown souce [sic].” The author claims that a “rapid rate of evolution” of the exon prevented its identification. Thus, the paper concludes: “For the moment we will posit that a genomic rearrangement (perhaps resulting from unequal crossing over) juxtaposed the first exon from an unknown donor gene to the 5′-flanking region of the ancestor of Adh-ψ.” The mutational odds of suddenly rearranging these stretches of DNA into one place to compose a functional gene are never considered.
Ignoring the warnings of AustinHughes, the author asserted, incredibly, that there was “rapid, adaptive evolution” and that “positive selection has played an important role in the evolution” of this gene even though the function of the gene is not known.
f. FOXP2
This gene is commonly cited as being involved in the origin of human language, even though it’s not exactly clear what it does.62 In fact, one study observed that “The finding that FOXP2 is critical to speech and language does not by itself demonstrate the role of this gene in the origin of human speech, because the function of FOXP2 could have remained unchanged during human evolution while other speech-related genes changed.”63
The studies cited by Long et al. compared human FOXP2 to copies of the same gene in chimps, gorillas, orangutans, the macaque, and mice, and found that “FOXP2 is a conserved protein, with only three amino acid differences (and a 1-amino-acid insertion/deletion) between human and mouse in its entire length of 715 amino acids.”64 Thus, this paper did not really study the origin of a new gene, but only tried to explain how human FOXP2 obtained a mere two differences in amino acid sequence from FOXP2 in apes.
In this case, the high ratio of non-synonymous (i.e. amino acid changing) to synonymous (i.e. silent) nucleotide differences was taken as evidence of the force of “positive selection.”65 Again, selection is being inferred, even though the authors didn’t know exactly what the gene does, violating Austin Hughes’s warning against “statistically based claim[s] of evidence for positive selection divorced from any biological mechanism.”66 At base, these studies catalogued interspecific differences between human FOXP2 and FOXP2 from other species, and found that those differences were extremely slight. Even if neo-Darwinian mechanisms were indeed at work, the degree of evolution in human FOXP2 amounts to 2 mutations, and 2 amino acid changes. This is an interesting finding, but not useful in explaining any actually noteworthy or impressive degrees of genetic evolution.
g. Cytochrome c1
This paper sought to explain the origin of a gene, cytochrome c1, involved in energy production in plants.67 The study found sequence similarity between three exons in cytochrome c1, a gene that operates in the mitochondria, with a gene with a very different function, GapC, which operates in the cytoplasm.68 That sequence similarity, essentially, formed the entire basis for this evolutionary story of rearrangement of exons, which made heavy use of Gene Evolution Game Rule 3. Since cytochrome c1 is less widespread than Gapc1, the authors concluded that Gapc1 is older and therefore “donated” the exons to cytochrome c1 through “exon shuffling.” Additionally, they speculate that the ancestral cytochrome c1 gene had the same function, but these new exons (for some reason) allowed the same function to be performed—but even more efficiently: “The ancestral cytochrome c1 gene in plants must have been targeted to the mitochondrion; thus this targeting sequence was replaced in the line leading to the potato by the GapC gene. This replacement may have been selected by some advantage in using the GapC promoter.” Predictably, the authors never discuss the mutational odds of replacing exons in one gene with exons “donated” from another gene such that the gene not only remains functional but has an advantage in performing its original function. This is the key phase where new genetic information must arise, but the authors never assess whether it would be likely to occur via unguided mutations.
h. Morpheus
This study aimed to explain the origin of a group of genes named morpheus that had changed so much that their origin could not be traced to any other gene. As the paper lamented, “some genes emerge and evolve very rapidly, generating copies that bear little similarity to their ancestral precursors” and thus “may not possess discernable orthologues within the genomes of model organisms.”69 When studying these genes, they reported “no significant sequence similarity to this gene family in other organisms at either the nucleotide or protein level.” Since it was impossible to invoke a scheme of duplications or other rearrangements from which this genetic material found its origin, the authors simply concluded, “These data suggested that the exonic regions were hypermutable or that amino-acid changes had been selected during the evolution of this gene family” and that their “analysis has revealed an extraordinary degree of evolutionary plasticity.” In other words, they have no idea where this gene came from, so they invoke the claim that the genes were “hypermutable” and subject to strong selection pressure such that their origin cannot be traced. How the genes actually arose is a question the authors never really address. Incredibly, they again appeal to strong selection pressure despite admitting “the precise function of this gene family is unknown.” Gene Evolution Game Rule 2 solved all the problems without anyone’s having to investigate the plausibility of the mechanism.
i. TRE2
This paper invoked “the chimeric fusion of two genes” to explain how the gene Tre2 evolved from duplicates of two other genes.70 The story is simple: Tre2 has 30 exons: exons 1-14 appear similar to another gene, TBCID3, while exons 15-30 are similar to the gene USP32. Thus the authors characterized the origin of this gene as “the abrupt creation of a mosaic gene with novel functions.” Although the authors claim that “domain accretion and gene-fusion events may not be uncommon,” they offered no consideration of the odds of mutations rearranging these two genes in a fashion that is functional and performs some new and useful function.
j. Dntf-2r
This study, co-authored by Long, claimed that Dntf-2r, a fruit fly gene, arose as a duplicate that was retrotransposed from the gene Dntf-2. Using Gene Evolution Game Rule 2, the authors attempt to explain the subsequent evolution of Dntf-2r by assessing the ratio of non-synonymous to synonymous differences. Using one test, they found that “polymorphism is higher for synonymous than for replacement sites … revealing the action of purifying selection,” however another test “revealed a significant excess of amino acid substitutions, suggesting that the accelerated protein sequence evolution is likely a consequence of the action of positive Darwinian selection.” To explain these seemingly contrary results, they decided that “both purifying selection and adaptive evolution” were at work. But they did not try to explain exactly what functions these forces were working to preserve or to change because the authors didn’t know the function of Dntf-2r. Before their study “there was no information on the function of Dntf-2r” and after their study, all they could say was “this gene may produce a functional protein.” Once again, positive selection is being conjured even though it is “divorced from any biological mechanism.”71 One would certainly like to know the mutational pathway taken or the selective advantage offered by specific mutations along that pathway. None of this is discussed, meaning an explanation for the evolution of new genetic information is absent from this paper.
The authors also tried to explain the origin of the promoter for Dntf-2r, rightly noting that “Whether or not a retroposed sequence recruits a new promoter is a critical step to its future fate. If a retroposed sequence integrates in a genomic region devoid of expression potential, it would be doomed to evolve into a pseudogene.” So how did Dntf-2r get its promoter? The authors found that Dntf-2r’s promoter fortuitously comes from DNA near where it’s located (its insertion site), but state that “it is unclear if this previously existing sequence is a functional promoter for some unknown gene in the region or is just a random genomic sequence that happens to be similar to a promoter sequence.”72 The authors make no attempt to assess the plausibility of these alternatives: they assess neither the likelihood of a “random genomic sequence” suddenly becoming a functional promoter sequence, nor the likelihood of a gene being inserted by chance right next to a functional promoter.
k. Sanguinaria rps1
This paper was inspired by the finding of “three striking distributional anomalies in a survey of mitochondrial gene content in angiosperms.”73 In other words, they found genes in species where they weren’t expected under the conventional understanding of common descent, because the same genes were found in supposedly “distantly related flowering plants.” Following Ragan and Beiko (“topological discordance between a gene tree and a trusted reference tree is taken as a prima facie instance of LGT [lateral gene transfer]”74), the authors assume that this phylogenetic incongruity is the result of LGT. This paper thus did not really explain the actual origin of these genes, but simply assumed and asserted that wherever and however they evolved, the genes were transplanted into these flowering plants via LGT (also known as horizontal gene transfer, or “HGT”).
The authors conclude that these data “establish for the first time that conventional genes are subject to evolutionarily frequent HGT during plant evolution and provide the first unambiguous evidence that plants can donate DNA horizontally to other plants.” Yet the authors admitted that the question “How do genes move from one plant to another, sexually unrelated, plant?” remains unanswered. Thus, evidence for HGT in plants is based merely upon the incongruent distribution of these genes assuming the standard phylogeny, not any actually established mechanism of HGT in flowering plants. Indeed, the authors admit that “horizontal transfer is unknown within the evolution of animals, plants and fungi except in the special context of mobile genetic elements.” This paper thus tells us virtually nothing about the actual original evolutionary birth of these genes, wherever they first originated, and instead highlights the assumptions and ad hoc reasoning used to save common descent from falsification by contrary phylogenetic data.
While studying this gene in various plant species, the authors found two additional instances of HGT, one of which was in Sanguinaria canadensis (bloodroot), a dicot whose rps11 gene “turns out to be chimaeric: its 5’ half is of expected eudicot, vertical origin, but its 3’ half is indisputably of monocot, horizontal origin.” In other words, half the gene appears like dicot rps11 and the other half appears like monocot rps11, and it is therefore identified as “chimaeric.” According to this story, monocot rps11 was transported into the Sanguinaria genome (by an unknown mechanism) and then, just by chance, happened to fuse with the dicot version of the same gene to create a new functional gene. The authors never discuss whether it is remotely plausible to claim that a gene would be transported from another species (by an unknown mechanism) only to fuse with its own homologue in the new genome—just by chance—and then create a new functional gene.
l. PMCHL
Despite the NCSE’s smooth assurance that “Biologists have no trouble showing how new information (in the sense used by information theorists) originates, nor how new genes, kinds of cells or tissues evolve,” this 2001 paper opens by admitting that “How genes with newly characterized functions originate remains a fundamental question.”75 Like the Sdic and AFGP examples, the origin of PMCHL1 and PMCHL2, considered here, required the “de novo” creation of key components of the gene where an exon “originated from a unique noncoding sequence.” The authors describe this process as requiring the “creation of 3’ exons from a unique noncoding genomic sequence that fortuitously evolved as a standard intron-exon structure and polyadenylation signal sequences.” Key portions of this gene therefore just “fortuitously evolved.” Is that an explanation? The paper does not want to encourage such arbitrary explanations, and thus the authors caution that “de novo generation of building blocks—single genes or gene segments coding for protein domains— seems to be rare.”
Accounting for the origin of the rest of this gene proved extremely complicated, but Gene Evolution Game Rules 1 and 3 allowed the authors to invoke a series of rearrangements including retrotranspositions, insertions, and duplications. They propose that these genes were suddenly “co-opted” or “‘exapted’ into a functional role.” While the origin of genes with new functions is indeed a “fundamental question,” this paper’s reliance on “fortuitously evolved” explanations does very little to answer that question. This is especially true considering that the authors offered no analysis of the mutational odds of converting noncoding DNA to coding DNA and recruiting and rearranging multiple segments of the genome to create a new functional gene.
VIII. Conclusion: The NCSE’s Citation Bluffs Reveal Little About the Evolutionary Origin of Information
More examples from that paper could be given, but the point is clear enough already: a careful analysis of Long et al. exposes the utterly insufficient explanations offered by neo-Darwinists to account for the origin of new genetic information.
In not a single case did the above papers cited by Long et al. actually explain how new functional information arose. In no case was there an analysis of how natural selection could have favored mutational changes that were shown to be likely along each step of an alleged evolutionary pathway; never was any detailed step-by-step mutational pathway even given. At best, these studies offered vague and ad hoc appeals to duplication, rearrangement, and natural selection — often in a sudden, extreme, and abrupt manner — to form the gene in question. In many cases, natural selection was invoked to allegedly account for changes in the gene, even though the investigators didn’t even know the function of the gene and thereby could not identify the advantage provided by the gene’s function. In no case were calculations performed to assess whether sufficient probabilistic resources existed to produce the asserted mutational events on a reasonable timescale. In some cases, the original genetic material for the genes was unknown, or the studies asserted spontaneous “de novo” origin of genes from previously non-coding DNA. While they readily admitted that “de novo” gene emergence is rare, no attempt was made to assess whether such an unguided mechanism is even remotely plausible on mathematical probabilistic grounds. These papers play the Gene Evolution Game, but never ask the right questions to explain how neo-Darwinian mechanisms create new genetic information.
The NCSE’s (and Judge Jones’s) citation bluffs have not explained how neo-Darwinian mechanisms produce new functional biological information. Instead, the mechanisms invoked in these papers are vague and hypothetical at best:
- exons may have been “recruited” or “donated” from other genes (and in some cases from an “unknown sou[r]ce”);
- there were vague appeals to “extensive refashioning of the genome”;
- mutations were said to cause “fortuitous juxtaposition of suitable sequences” in a gene-promoting region that therefore “did not really ‘evolve’”;
- researchers assumed “radical change in the structure” due to “rapid, adaptive evolution” and claimed that “positive selection has played an important role in the evolution” of the gene, even though function of the gene was not even known;
- genes were purportedly “cobbled together from DNA of no related function (or no function at all)”;
- the “creation” of new exons “from a unique noncoding genomic sequence that fortuitously evolved” was assumed, not demonstrated;
- we were given alternatives that promoter regions arose from a “random genomic sequence that happens to be similar to a promoter sequence,” or that the gene arose because it was inserted by pure chance right next to a functional promoter.
- explanations went little further than invoking “the chimeric fusion of two genes” based solely on sequence similarity;
- when no source material is recognizable, we’re told that “genes emerge and evolve very rapidly, generating copies that bear little similarity to their ancestral precursors” because they are simply “hypermutable”;
- we even saw “a striking case of convergent evolution” of “near-identical” proteins.
To reiterate, in no cases were the odds of these unlikely events taking place actually calculated. Incredibly, natural selection was repeatedly invoked in instances where the investigators did not know the function of the gene being studied and thus could not possibly have identified any known functional advantages gained through the mutations being invoked. In the case where multiple mutational steps were involved, no tests were done of the functional viability of the alleged intermediate stages. These papers offer vague stories but not viable, plausibly demonstrated explanations for the origin of new genetic information.
Within modern evolutionary biology, there are indeed many unanswered questions about how unguided selection acting upon random mutation produces new functional biological information. This will be the legacy of the Kitzmiller ruling: It pretends that fundamental scientific questions have been answered, when no adequate answers exist, at least not from the neo-Darwinian paradigm. As a result, the approach taken by Judge Jones and his consorts at the NCSE not only would miseducate students, but it threatens to hinder scientific progress by pretending that some of the most important questions in biology are answered, when they really aren’t. The myth unfortunately has become a pillar of how evolution is explained and defended in law, academia, education and the media.
References Cited:
[1.] Nicholas Matzke quoted in Michael Powell, “Controversial Editor Backed,” Washington Post (August 19, 2005).
[2.] Kenneth R. Miller, Kitzmiller v. Dover Day 1 AM testimony, pg. 135 (September 26, 2005).
[3.] Kitzmiller v. Dover, 400 F.Supp.2d 707, 744 (M.D.Pa. 2005).
[4.] Manyuan Long, Esther Betrán, Kevin Thornton, and Wen Wang, “The Origin of New Genes: Glimpses from the Young and Old,” Nature Reviews Genetics, Vol. 4:865-875 (November, 2003).
[5.] The word “information” appears once in the entire article—in the title of note 103. Id. at 875 n. 103. See Manyuan Long, Esther Betrán, Kevin Thornton, and Wen Wang, “The Origin of New Genes: Glimpses from the Young and Old,” Nature Reviews Genetics, Vol. 4:865-875 (November, 2003).
[6.] Nicholas J. Matzke and Paul R. Gross, “Analyzing Critical Analysis: The Fallback Antievolutionist Strategy,” pg. 42 in Not in Our Classrooms: Why Intelligent Design is Wrong for Our Schools (edited by Eugenie C. Scott and Glenn Branch, Beacon Press, 2006).
[7.] Id.
[8.] Bernd-Olaf Kuppers, Information and the Origin of Life (Cambridge: MIT Press, 1990), pp. 170–172.
[9.] This calculation uses a 26 letter English alphabet that is not case-sensitive and, as seen in the strings, does not use spaces.
[10.] String B was generated using a random character generator from the website Random.org.
[11.] Jack W. Szostak, “Molecular messages,” Nature, Vol. 423:689 (June 12, 2003).
[12.] Kirk K. Durston, David K. Y. Chiu, David L. Abel, Jack T. Trevors, “Measuring the functional sequence complexity of proteins,” Theoretical Biology and Medical Modelling, Vol. 4:47 (2007) (internal citations removed).
[13.] Robert M. Hazen, Patrick L. Griffin, James M. Carothers, and Jack W. Szostak, “Functional information and the emergence of biocomplexity,” Proceedings of the National Academy of Sciences, USA, Vol. 104:8574–8581 (May 15, 2007).
[14.] Stephen C. Meyer, “The origin of biological information and the higher taxonomic categories,” Proceedings of the Biological Society of Washington, Vol. 117(2):213-239 (2004).
[15.] Leslie E. Orgel, The Origins of Life: Molecules and Natural Selection, pg. 189 (Chapman & Hall: London, 1973).
[16.] Comment by Michael Egnor at http://scienceblogs.com/pharyngula/2007/02/dr_michael_egnor_challenges_ev.php#comment-349555 (February 20, 2007)
[17.] Again, as implied in the body, if one could predict the string would be duplicated, then the Shannon Information would also not increase after duplicating the string, in which case there is no increase in CSI nor Shannon Information.
[18.] Michael Lynch, “The frailty of adaptive hypotheses for the origins of organismal complexity,” Proceedings of the National Academy of Sciences, Vol. 104:8597–8604 (May 15, 2007).
[19.] Austin L. Hughes, “The origin of adaptive phenotypes,” Proceedings of the National Academy of Sciences USA, Vol. 105(36):13193–13194 (Sept. 9, 2008) (internal citations removed).
[20.] Michael J. Behe, Darwin’s Black Box: The Biochemical Challenge to Evolution, pgs. 175-176 (Free Press, 1996).
[21.] Michael J. Behe, The Edge of Evolution: The Search for the Limits of Darwinism, pg. 95 (Free Press, 2007).
[22.] See for example, “Limits on Evolution” at http://ncseweb.org/creationism/analysis/extrapolations
[23.] For example, when the ratio of nonsynonymous (i.e. amino acid changing) to synonymous (i.e. non-amino acid changing) differences between Gene B and Gene A is high, we have can say that it must be natural selection at work because only strong selection pressure would preserve so many changes that change amino acid sequence. Incredibly, we can also say that when the same ratio is low (i.e. there are FEW amino acid replacements in a gene), that too shows that natural selection was at work, in this case in the form of stabilizing selection to conserve gene sequence. This approach was taken in Harmit S. Malik and Steven Henikoff, “Adaptive Evolution of Cid, a Centromere-Specific Histone in Drosophila,” Genetics, Vol. 157:1293–1298 (March 2001) and its discussion of the Cid gene in a subsequent post.
[24.] See for example Ulfar Bergthorsson, Keith L. Adams, Brendan Thomason, and Jeffrey D. Palmer, “Widespread horizontal transfer of mitochondrial genes in flowering plants,” Nature, Vol. 424:197-201 (July 10, 2003). See also Mark A. Ragan and Robert G. Beiko, “Lateral genetic transfer: open issues,” Philosophical Transactions of the Royal Society B, Vol. 364:2241-2251 (2009) (“topological discordance between a gene tree and a trusted reference tree is taken as a prima facie instance of LGT”).
[25.] For example, this explanation was invoked in Matthew E. Johnson, Luigi Viggiano, Jeffrey A. Bailey, Munah Abdul-Rauf, Graham Goodwin, Mariano Rocchi & Evan E. Eichler, “Positive selection of a gene family during the emergence of humans and African apes,” Nature, Vol. 413:514-519 (October 4, 2001).
[26.] Austin L. Hughes, “Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level,” Heredity, Vol. 99:364–373 (2007).
[27.] Austin L. Hughes, “The origin of adaptive phenotypes,” Proceedings of the National Academy of Sciences USA, Vol. 105(36):13193–13194 (Sept. 9, 2008).
[28.] Id.
[29.] “The modern synthesis is good at modeling the survival of the fittest, but not the arrival of the fittest.” Scott Gilbert, quoted in John Whitfield, “Biological Theory: Postmodern evolution?,” Nature, Vol. 455:281-284 (2008).
[30.] Bernard Wood, quoted in Joseph B. Verrengia, “Gene Mutation Said Linked to Evolution,” Associated Press, found in San Diego Union Tribune, March 24, 2004.
[31.] Jerry Coyne, “The Great Mutator,” The New Republic (June 14, 2007). Coyne asserts he knows of no example where this is the case.
[32.] David Berlinski, “Keeping an Eye on Evolution: Richard Dawkins, a relentless Darwinian spear carrier, trips over Mount Improbable. Review of Climbing Mount Improbable by Richard Dawkins (W. H. Norton & Company, Inc. 1996),” in The Globe & Mail (November 2, 1996) at http://www.discovery.org/a/132
[33.] Douglas A. Axe, “Estimating the Prevalence of Protein Sequences Adopting Functional Enzyme Folds,” Journal of Molecular Biology, Vol. 341: 1295-1315 (2004); Douglas A. Axe, “Extreme Functional Sensitivity to Conservative Amino Acid Changes on Enzyme Exteriors,” Journal of Molecular Biology, Vol. 301: 585-595 (2000).
[34.] See Stephen C. Meyer, Signature in the Cell: DNA and the Evidence for Intelligent Design, pg. 211 (Harper One, 2009).
[35.] Neil A. Campbell and Jane B. Reece, Biology, pg. 84 (7th ed, 2005).
[36.] David S. Goodsell, The Machinery of Life, pg. 17, 19 (2nd ed, Springer, 2009).
[37.] Michael J. Behe & David W. Snoke, “Simulating Evolution by Gene Duplication of Protein Features That Require Multiple Amino Acid Residues,” Protein Science, Vol 13:2651-2664 (2004).
[38.] Rick Durrett and Deena Schmidt, “Waiting for Two Mutations: With Applications to Regulatory Sequence Evolution and the Limits of Darwinian Evolution,” Genetics, Vol. 180: 1501–1509 (November 2008).
[39.] Michael Behe, The Edge of Evolution: The Search for the Limits of Darwinism, Appendix D, pgs. 272-275 (Free Press, 2007) (emphasis added).
[40.] Austin L. Hughes, “The origin of adaptive phenotypes,” Proceedings of the National Academy of Sciences USA, Vol. 105(36):13193–13194 (Sept. 9, 2008) (internal citations removed).
[41.] Michael Lynch, “The frailty of adaptive hypotheses for the origins of organismal complexity,” Proceedings of the National Academy of Sciences, Vol. 104:8597–8604 (May 15, 2007).
[42.] Manyuan Long, Esther Betrán, Kevin Thornton, and Wen Wang, “The Origin of New Genes: Glimpses from the Young and Old,” Nature Reviews Genetics, Vol. 4:865-875 (November, 2003).
[43.] Kitzmiller v. Dover, 400 F.Supp.2d 707, 744 (M.D.Pa. 2005).
[44.] See Limits on Evolution at http://ncseweb.org/creationism/analysis/extrapolations
[45.] Manyuan Long & Charles H. Langley, “Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila,” Science, Vol. 260:91–95 (April 2, 1993).
[46.] John M. Logsdon, Jr., & W. Ford Doolittle, “Origin of antifreeze protein genes: A cool tale in molecular evolution,” Proceedings of the National Academy of Sciences USA, Vol. 94:3485-3487 (April, 1997).
[47.] Austin L. Hughes, “Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level,” Heredity, Vol. 99:364–373 (2007).
[48.] Dmitry I. Nurminsky, Maria V. Nurminskaya, Daniel De Aguiar, and Daniel L. Hartl, “Selective sweep of a newly evolved sperm-specic gene in Drosophila,” Nature, Vol. 396:572-575 (December 10, 1998).
[49.] Id.
[50.] Manyuan Long, Esther Betrán, Kevin Thornton, and Wen Wang, “The Origin of New Genes: Glimpses from the Young and Old,” Nature Reviews Genetics, Vol. 4:865-875 (November, 2003).
[51.] Harmit S. Malik and Steven Henikoff, “Adaptive Evolution of Cid, a Centromere-Specific Histone in Drosophila,” Genetics, Vol. 157:1293–1298 (March 2001).
[52.] Austin L. Hughes, “The origin of adaptive phenotypes,” Proceedings of the National Academy of Sciences USA, Vol. 105(36):13193–13194 (Sept. 9, 2008) (internal citations removed).
[53.] Liangbiao Chen, Arthur L. DeVries, & Chi-Hing C. Cheng, “Convergent evolution of antifreeze glycoproteins in Antarctic notothenioid fish and Arctic cod,” Proceedings of the National Academy of Sciences USA, Vol. 94:3817–3822 (April, 1997).
[54.] John M. Logsdon, Jr., & W. Ford Doolittle, “Origin of antifreeze protein genes: A cool tale in molecular evolution,” Proceedings of the National Academy of Sciences USA, Vol. 94:3485-3487 (April, 1997).
[55.] Id.
[56.] Id.
[57.] Liangbiao Chen, Arthur L. DeVries, & Chi-Hing C. Cheng, “Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish,” Proceedings of the National Academy of Sciences USA, Vol. 94:3811–3816 (April, 1997).
[58.] John M. Logsdon, Jr., & W. Ford Doolittle, “Origin of antifreeze protein genes: A cool tale in molecular evolution,” Proceedings of the National Academy of Sciences USA, Vol. 94:3485-3487 (April, 1997).
[59.] Liangbiao Chen, Arthur L. DeVries, & Chi-Hing C. Cheng, “Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish,” Proceedings of the National Academy of Sciences USA, Vol. 94:3811–3816 (April, 1997).
[60.] John M. Logsdon, Jr., & W. Ford Doolittle, “Origin of antifreeze protein genes: A cool tale in molecular evolution,” Proceedings of the National Academy of Sciences USA, Vol. 94:3485-3487 (April, 1997).
[61.] David J. Begun, “Origin and Evolution of a New Gene Descended From alcohol dehydrogenase in Drosophila,” Genetics, Vol. 145:375-382 (February, 1997).
[62.] Wolfgang Enard, Molly Przeworski, Simon E. Fisher, Cecilia S. L. Lai, Victor Wiebe, Takashi Kitano, Anthony P. Monaco & Svante Pääbo, “Molecular evolution of FOXP2, a gene involved in speech and language,” Nature, Vol. 418:869-872 (August 22, 2002) (stating “to establish whether FOXP2 is indeed involved in basic aspects of human culture, the normal functions of both the human and the chimpanzee FOXP2 proteins need to be clarified”).
[63.] Jianzhi Zhang, David M. Webb and Ondrej Podlaha, “Accelerated Protein Evolution and Origins of Human-Specific Features: FOXP2 as an Example,” Genetics, Vol. 162:1825–1835 (December 2002).
[64.] Id.
[65.] Id.
[66.] Austin L. Hughes, “Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level,” Heredity, Vol. 99:364–373 (2007).
[67.] Manyuan Long, Sandro J. de Souza, Carl Rosenberg, and Walter Gilbert, “Exon shuffling and the origin of the mitochondrial targeting function in plant cytochrome cl precursor,” Proceedings of the National Academy of Sciences USA, Vol. 93:7727-7731 (July, 1996).
[68.] Id. Specifically, the authors write: “In a computer survey of an exon database, we observed a high similarity (44% identity and 64% similarity over 41 amino acids) between the 5’ three consecutive exons of the pea Gapc1 and the potato cytochrome c1 precursor.”
[69.] Matthew E. Johnson, Luigi Viggiano, Jeffrey A. Bailey, Munah Abdul-Rauf, Graham Goodwin, Mariano Rocchi & Evan E. Eichler, “Positive selection of a gene family during the emergence of humans and African apes,” Nature, Vol. 413:514-519 (October 4, 2001).
[70.] Charles A. Paulding, Maryellen Ruvolo, and Daniel A. Haber, “The Tre2 (USP6) oncogene is a hominoid-specific gene,” Proceedings of the National Academy of Sciences USA, Vol. 100(5):2507–2511 (March 4, 2003).
[71.] Austin L. Hughes, “Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level,” Heredity, Vol. 99:364–373 (2007).
[72.] Esther Betran and Manyuan Long, “Dntf-2r, a Young Drosophila Retroposed Gene With Specific Male Expression Under Positive Darwinian Selection,” Genetics, Vol. 164:977–988 ( July 2003).
[73.] Ulfar Bergthorsson, Keith L. Adams, Brendan Thomason, and Jeffrey D. Palmer, “Widespread horizontal transfer of mitochondrial genes in flowering plants,” Nature, Vol. 424:197-201 (July 10, 2003).
[74.] Mark A. Ragan and Robert G. Beiko, “Lateral genetic transfer: open issues,” Philosophical Transactions of the Royal Society B, Vol. 364:2241-2251 (2009).
[75.] Anouk Courseaux and Jean-Louis Nahon, “Birth of Two Chimeric Genes in the Hominidae Lineage,” Science, Vol. 291:1293-1297 (February 16, 2001).